We use cookies and other tracking technologies to improve your browsing experience on our website, to show you personalized content and targeted ads, to analyze our website traffic, and to understand where our visitors are coming from.
The Distribution Concept
Published
August 24, 2025
Under construction
The pharmacokinetic parameter that quantifies drug distribution, is called volume of distribution ().
In general, a lipophilic drug will have a large , and a hydrophilic drug will have a small . Drugs can also be substrates to various transporters that impact .
usually has 30% inter-individual variability.
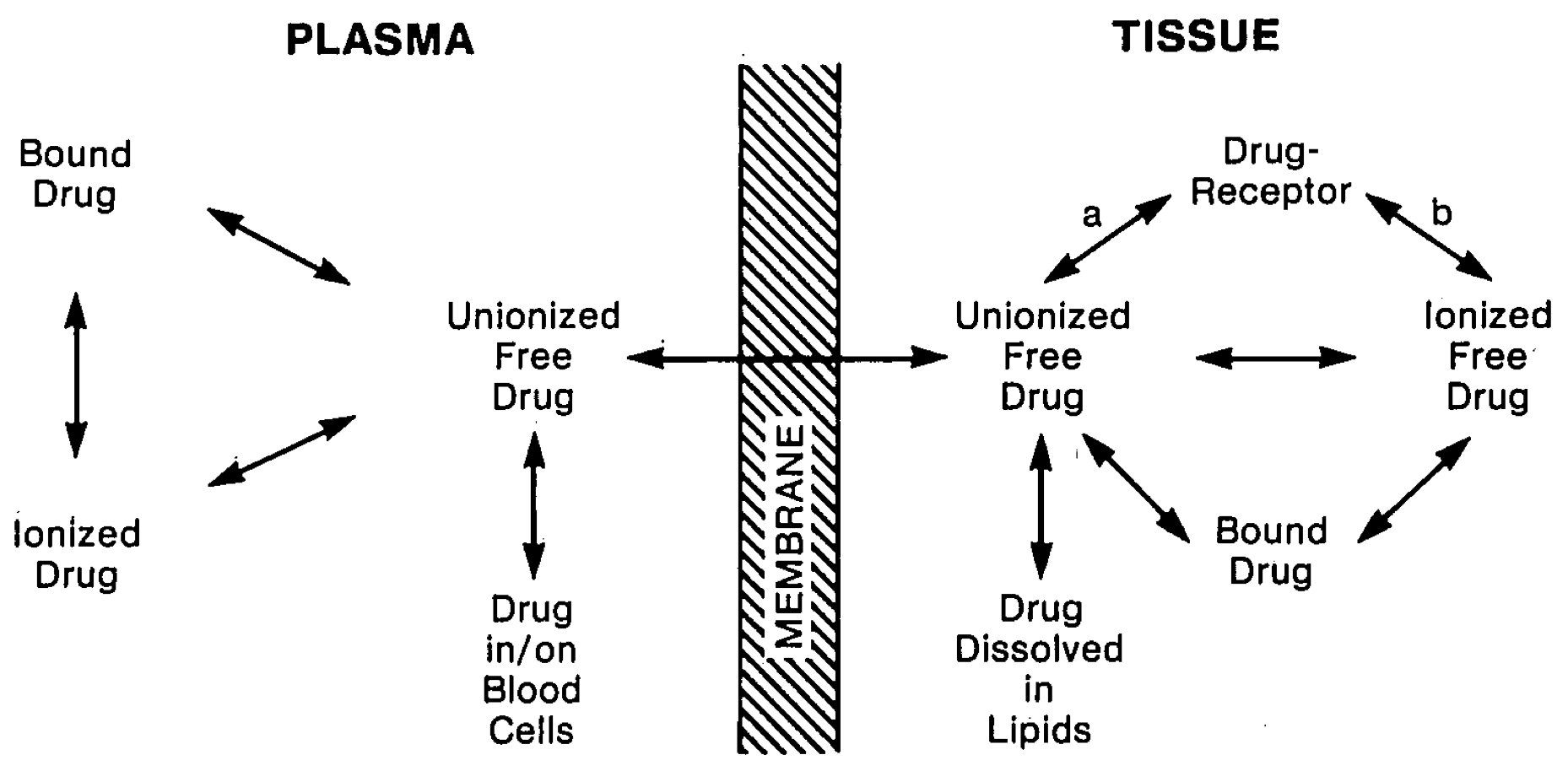
Figure 1: Reversible processes affecting the total amount of drug on each side of a biological membrane permeable only to the un-ionized free form of a drug. Some binding of both the un-ionized and ionized forms may occur, but depending on the specific drug, one form usually predominates (the un-ionized form is preferentially bound in the case of narcotic analgesics). Similarly, at active receptor sites, one form of the drug usually is effective and the other is not (a or b). A very high affinity of the drug for pharmacologically inactive binding sites in tissues or a large partitioning of a lipophillic drug into lipids creates a sink for drug accumulation in tissue and limits the free drug concentration, which determines the number of drug-receptor complexes formed and the intensity of drug action. Extensive binding and partitioning in tissue creates a large tissue distribution volume.